Surface d’une molécule
Démarche proposée
- Définir la ou les surfaces d’une molécule et ce que l’on cherche à calculer dans un premier temps
- Analyser un fichier PDB.
- Définir une méthode de calcul.
- Implémenter un algorithme et le tester sur des molécules de tailles variables.
- Comparer la méthode à des programmes existants.
- Évaluer l’efficacité de l’algorithme et identifier les principales contraintes liées au temps de calcul.
Surface d’une molécule : définitions
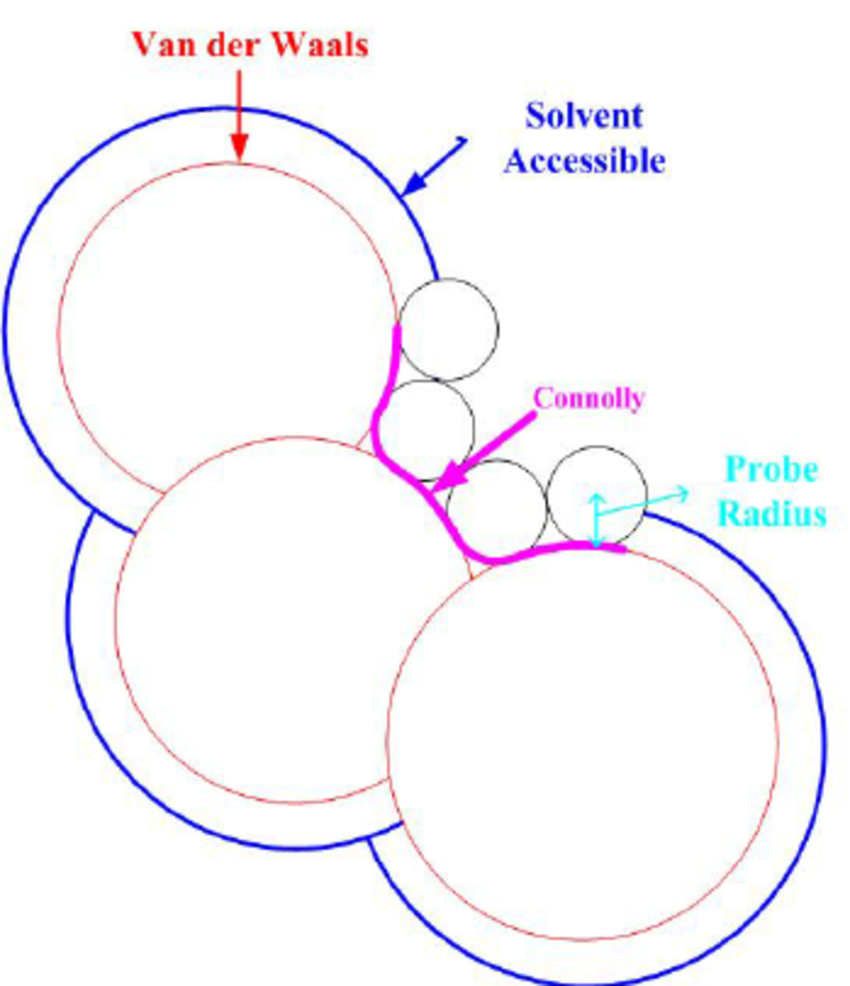
Figure 1 : Défition des surfaces de molécule : surface de Van der Waals(VDW), surface accessible au solvent (SAS) et surface exclue au solvent (SES) ou surface de Connely. Source : Sourina2009
Discrétisation totale de l’espace 3D
Pour calculer des propriétés géométriques comme le volume ou la surface de l’intersection des sphères de VDW, on peut utiliser une méthode basée sur des grilles. On cherche alors à représenter la densité des atomes sur une grille discrète. La taille de la grille définit la résolution du système.
En C, on pourra utiliser, par exemple un tableau tridimensionnel prédéfini au début du programme :
// Taille de la grille en X, Y et Z (ajustez-la en fonction de votre besoin)
#define GRID_SIZE_X 100
#define GRID_SIZE_Y 100
#define GRID_SIZE_Z 100
// Fonction pour initialiser la grille
void initializeGrid(int grid[GRID_SIZE_X][GRID_SIZE_Y][GRID_SIZE_Z]) {
for (int x = 0; x < GRID_SIZE_X; x++) {
for (int y = 0; y < GRID_SIZE_Y; y++) {
for (int z = 0; z < GRID_SIZE_Z; z++) {
grid[x][y][z] = 0; // Initialise toutes les cellules à 0
}
}
}
}
On pourra améliorer cette approche en introduisant un pas de la grille et en tenant compte des dimensions de la molécule étudiée.
La densité d’atomes peut être représentée sur une grille, et vaut 0 à l’extérieur de la sphère de VDW et 1 à l’intérieur. Un algorithme de détection de surface à partir de la densité est nécessaire pour déterminer les points situés sur la surface externe des sphères de VDW.
Approche avec discrétisation partielle de l’espace
Dans la méthode de Lee & Richards, l’espace est discrétisé seulement suivant une direction de l’espace, par exemple suivant z. Cela revient à diviser la molécule en tranches d’épaisseur $\delta$. Pour chaque tranche, l’intersection d’une sphère de VDW avec le plan horizontal est un cercle. Il convient ensuite de calculer la contribution de chaque arc de cercle dirigé vers l’extérieur de la molécule.
Liens utiles :
- documentation du code FreeSASA
- Article historique : Lee, B.; Richards, F. M. The Interpretation of Protein Structures: Estimation of Static Accessibility. Journal of Molecular Biology 1971, 55 (3), 379-IN4. https://doi.org/10.1016/0022-2836(71)90324-X.
Méthode numérique de Shrake-Rupley
Cette méthode utilise également la notion de sphère sonde roulant autour d’une molécule, mais utilise des points répartis de manière équidistante autour de chaque atome. L’algorithme parcourt chaque point et vérifie s’il est interne (vers l’intérieur de la molécule) ou externe (vers l’extérieur de la molécule).
Liens utiles :
- page wikipédia
- Article historique : Shrake, A.; Rupley, J. A. Environment and Exposure to Solvent of Protein Atoms. Lysozyme and Insulin. Journal of Molecular Biology 1973, 79 (2), 351–371. https://doi.org/10.1016/0022-2836(73)90011-9.
Méthodes purement analytiques
Il est possible de calculer analytiquement, c’est-à-dire à l’aide de formulaires exactes les surfaces d’une molécule en utilisant une approche géométrique du problème. Cela correspond essentiellement à déterminer la surface interne des intersections de paires de sphères et les exclure de la surface totale générée par le roulage d’une sphère sonde autour de chaque atome.
Liens utiles :
- Article récent, complet, avec une approche mathématique : Quan, C.; Stamm, B. Mathematical Analysis and Calculation of Molecular Surfaces. Journal of Computational Physics 2016, 322, 760–782. https://doi.org/10.1016/j.jcp.2016.07.007.
Optimisation
Certaines étapes de calcul peuvent être limitantes. On pourra utiliser des librairies comme KDTree qui permettent de réduire la coût de calcul dans la recherche de plus proches voisins.
Liens utiles :
- Page wikipédia Arbre kd
- une librairie KDtree en C
Quelques programmes déjà existants :
Il existe de nombreux codes pour calculer les différentes surfaces de molécules biologiques. En général, les codes sont complexes car ils proposent des options supplémentaires, traitent les cas difficiles et sont optimisés pour leur performance. Cela peut être toutefois intéressant de comparer les résultats des différents codes. Voici une liste non exhaustive de ces codes ou logiciels contenant des librairies de calcul de surface :